FACS Antibodies
Fluorescence-activated cell sorting (FACS) is a specialized type of flow cytometry. It sorts a heterogeneous mixture of biological cells into two or more containers, one cell at a time, based upon the specific light scattering and fluorescent characteristics of each cell and its attached antibodies.
Important Targets
All Flow Cytometry Antibodies
Recommended next Filters:
105,277 results
Independently Validated
ACTB
适用: 人, 小鼠, 大鼠, 兔, Pig, Cow
WB, ELISA, IHC (p), FACS, IF (p), IF (cc)
宿主: 兔
Polyclonal
unconjugated
 272 references
272 references
Rockland Original
Rockland Original
Rockland 200-401-a50
SARS-CoV N
适用: SARS Coronavirus (SARS-CoV), SARS Coronavirus-2 (SARS-CoV-2)
ELISA, WB, IHC, FACS, FM
宿主: 兔
Polyclonal
unconjugated
97 references
Rockland Original
Rockland 600-401-103-0.1
适用: Cow, 人, 小鼠, Pig, 大鼠
DB, ELISA, FLISA, FACS, FM, IHC, IP, WB
宿主: 兔
Polyclonal
unconjugated
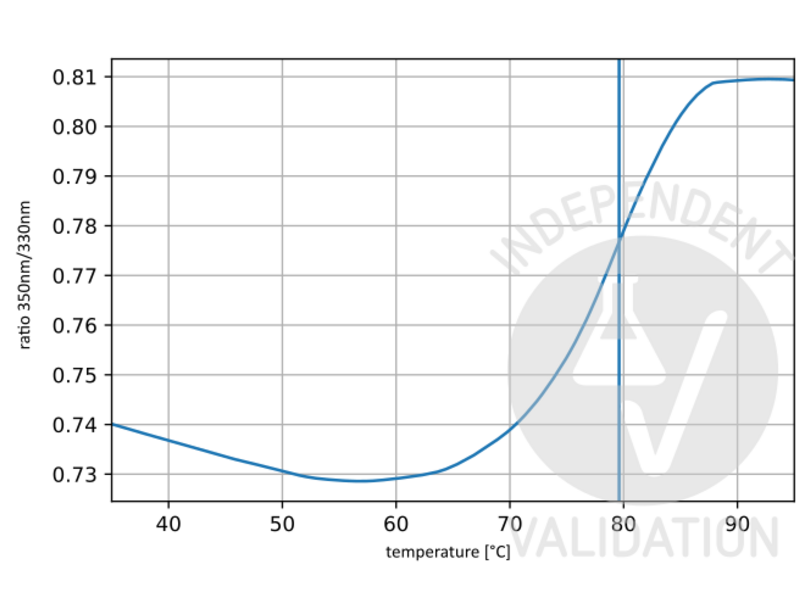
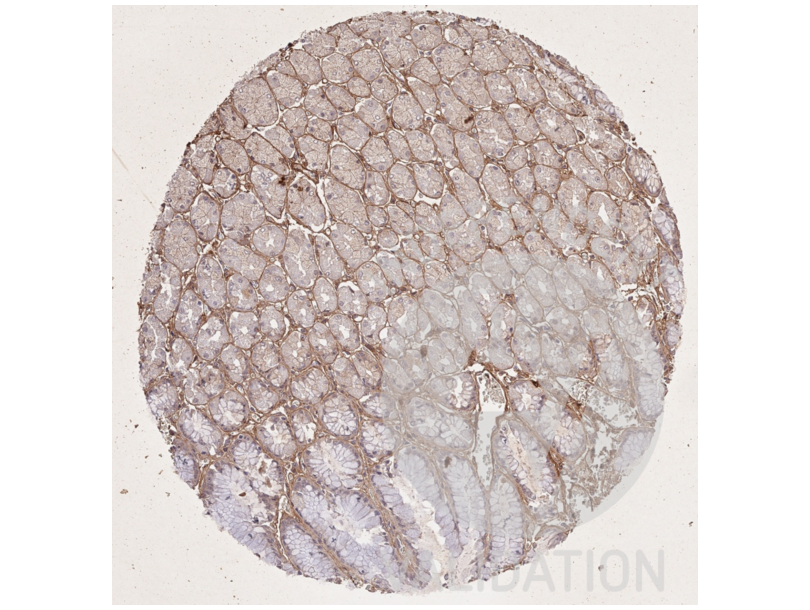
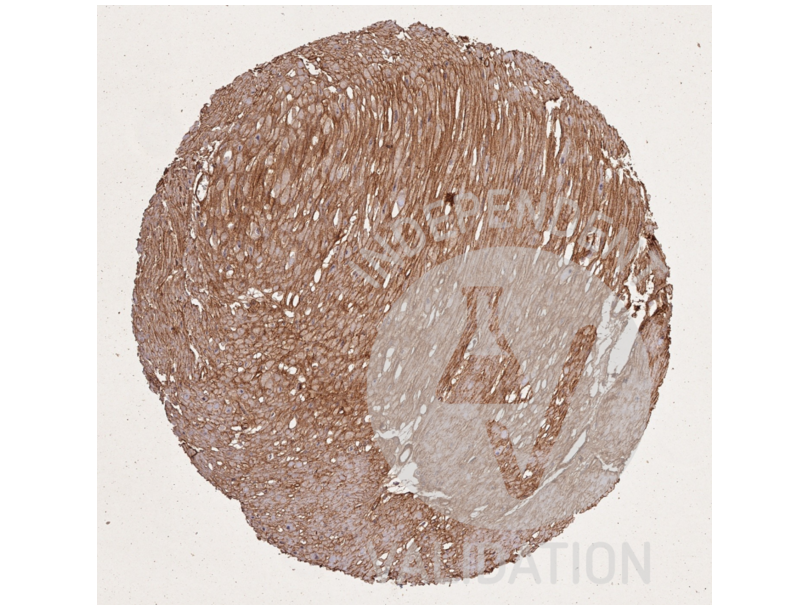
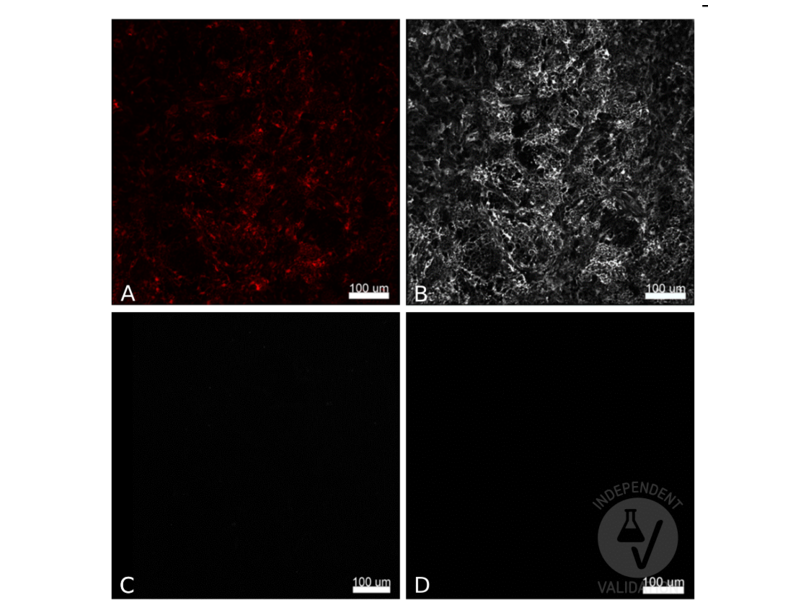
 63 references
63 references
 2 validations
2 validations
Independently Validated
NFkBP65
适用: 人, 小鼠, 大鼠
WB, ELISA, IF, FACS, ICC
宿主: 兔
Polyclonal
unconjugated
45 references
Rockland Original
You are here:



